Что такое спинальная мышечная атрофия?
Спинальная мышечная атрофия (СМА) — это группа наследственных заболеваний, которые постепенно разрушают двигательные нейроны — нервные клетки в стволе головного и спинного мозга, которые контролируют основную активность скелетных мышц, такую как речь, ходьба, дыхание и глотание, что приводит к мышечной слабости и атрофии. Двигательные нейроны контролируют движения в руках, ногах, груди, лице, горле и языке. Когда происходят сбои в сигналах между двигательными нейронами и мышцами, мышцы постепенно ослабевают, начинают истощаться и развиваются подергивания, их называют фасцикуляциями.
Что вызывает СМА?
Наиболее распространенная форма СМА вызвана дефектами в обеих копиях гена выживания моторного нейрона 1 (SMN1) на хромосоме 5q. Этот ген продуцирует белок выживания моторного нейрона (SMN), который поддерживает здоровье и нормальную функцию моторных нейронов. Люди с СМА имеют недостаточный уровень белка SMN, что приводит к потере двигательных нейронов в спинном мозге, вызывая слабость и истощение скелетных мышц. Эта слабость часто более выражена в мышцах туловища и верхней части ног и предплечий, чем в мышцах кистей и стоп.
Существует много типов спинальной мышечной атрофии, которые вызваны изменениями в одних и тех же генах. Менее распространенные формы СМА вызваны мутациями в других генах, включая ген VAPB, расположенный на хромосоме 20, ген DYNC1H1 на хромосоме 14, ген BICD2 на хромосоме 9 и ген UBA1 на Х-хромосоме. Типы различаются по возрасту начала и тяжести мышечной слабости.
Примерно 95-98% пораженных людей имеют делеции в гене SMN1, а 2-5% имеют точечную мутацию в гене SMN1, которая приводит к снижению выработки белка SMN.
Ген SMN2 является паралогом SMN1 и также кодирует белок SMN и может частично компенсировать потерю гена SMN1. Однако большая часть белка SMN, продуцируемого геном SMN2, не является функциональной, поэтому ген SMN2 может лишь частично компенсировать потерю гена SMN1. По этой причине человек с СМА, у которого больше копий гена SMN2, будет вырабатывать более функциональный белок SMN и, возможно, будет лучше способен компенсировать потерю гена SMN1, что, следовательно, приведет к менее тяжелому заболеванию. Как правило, большее количество копий SMN2 ассоциировано с более легким заболеванием СМА, хотя есть исключения.
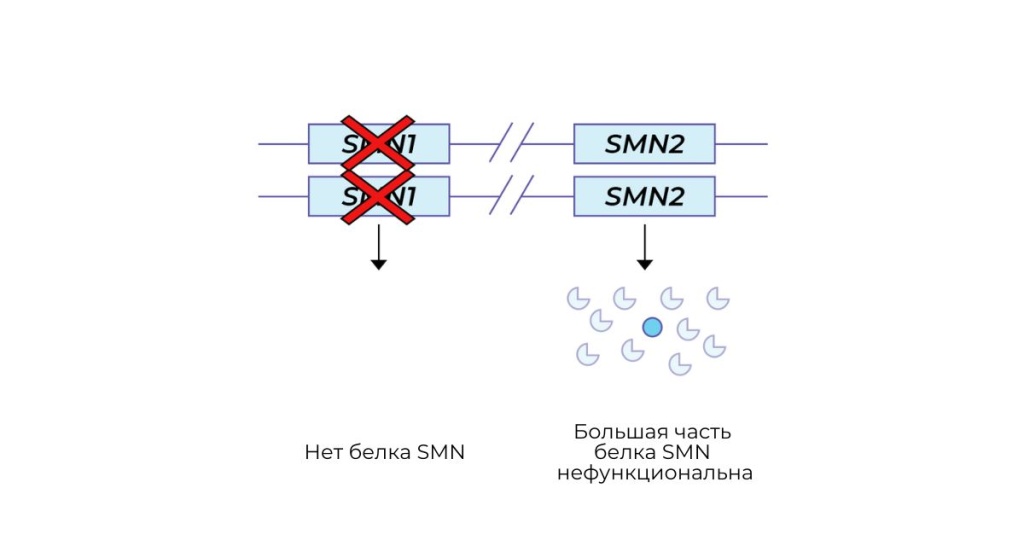
Разница между геном CMN1 и геном CMN2
Как СМА передается по наследству?
За исключением редких случаев, вызванных мутациями в гене UBA1, SMA наследуется аутосомно-рецессивным способом, что означает, что пораженный индивидуум имеет два мутировавших гена, часто наследуя по одному от каждого родителя. Те, кто несет только один мутировавший ген, являются носителями заболевания без каких-либо симптомов. Аутосомно-рецессивные заболевания могут поражать более одного человека в одном поколении (братьев, сестер или двоюродных братьев).
Риск того, что оба родителя-носителя передадут вариантный ген и, следовательно, родят больного ребенка, составляет 25% при каждой беременности. Риск рождения ребенка, который является носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей, составляет 25%. Риск одинаков для мужчин и женщин.
Люди, у которых больше, чем обычно, двух копий гена SMN2, обычно не наследуют дополнительные копии от родителя. Обычно они возникают в результате случайной ошибки при создании новых копий ДНК (репликации) в яйцеклетке или сперматозоиде или сразу после оплодотворения.
Каковы типы СМА?
Существует широкий спектр нарушений, наблюдаемых при СМА, вызванных дефектами в гене SMN1, от возникновения до рождения с затрудненным дыханием при рождении до легкой слабости у взрослых. Соответственно, эта наиболее распространенная форма СМА может быть классифицирована на четыре типа в зависимости от достигнутого наивысшего двигательного порога.
СМА I типа, также называемая болезнью Верднига-Хоффмана или СМА с младенческим началом, проявляется обычно в возрасте до 6 месяцев. Наиболее серьезно пораженные младенцы (СМА типа 0 или IA) имеют ограниченные движения даже внутриутробно и рождаются с контрактурами и затрудненным дыханием, причем смерть наступает на первом году жизни без лечения. Симптомы СМА I типа включают гипотонию (снижение мышечного тонуса), уменьшение движений конечностей, отсутствие сухожильных рефлексов, фасцикуляций, трудности с глотанием и кормлением, а также нарушение дыхания. У этих детей также с возрастом развивается сколиоз (искривление позвоночника) или другие аномалии скелета. Без какого-либо лечения пораженные дети никогда не сидят и не стоят, и подавляющее большинство обычно умирают от дыхательной недостаточности в возрасте до 2 лет. Дети с СМА I типа теперь живут дольше и могут достичь более высоких двигательных показателей, таких как сидение и даже ходьба, благодаря более активному клиническому уходу и новому доступному лечению.
Дети с СМА II типа, промежуточной формой, обычно проявляют свои первые симптомы в возрасте от 6 до 18 месяцев, хотя у некоторых они могут проявляться и раньше. Они способны сидеть без поддержки, но не в состоянии стоять или ходить без посторонней помощи, а некоторые могут со временем потерять способность самостоятельно сидеть без лечения. У них могут быть проблемы с дыханием, включая гиповентиляцию во сне. Прогрессирование заболевания изменчиво без лечения. Ожидаемая продолжительность жизни сокращается, но большинство людей доживают до подросткового или юношеского возраста. Благодаря лечению, модифицирующему заболевание, и активному клиническому уходу у детей с СМА II типа улучшились двигательные показатели.
У детей с СМА III типа (болезнь Кугельберга-Веландера) симптомы развиваются после 18 месяцев, и они могут самостоятельно ходить. Сначала они испытывают трудности при ходьбе и беге, подъеме по ступенькам или вставании со стула. В первую очередь чаще всего поражаются проксимальные мышцы ног, при этом в руках наблюдается тремор. Осложнения включают сколиоз и контрактуры суставов — хроническое укорочение мышц или сухожилий вокруг суставов, вызванное ненормальным мышечным тонусом и слабостью, которые препятствуют свободному движению суставов. Люди с СМА III типа могут быть склонны к респираторным инфекциям, но при соблюдении осторожности большинство из них имеют нормальную продолжительность жизни. Лечение, модифицирующее заболевание, может улучшить результаты.
У людей с СМА IV типа симптомы развиваются после 21 года, с легкой или умеренной слабостью проксимальных мышц и другими симптомами.
Как диагностируется СМА?
Для выявления делеций или мутаций гена SMN1 можно сдать анализ крови. Этот тест выявляет по меньшей мере 95 процентов типов СМА I, II и III, а также может выявить, является ли человек носителем дефектного гена, который может быть передан детям. Если ген SMN1 не обнаружен аномальным или история болезни и обследование пациента не типичны для СМА, другие диагностические тесты могут включать электромиографию (которая регистрирует электрическую активность мышц во время сокращения и в состоянии покоя), исследования скорости нервной проводимости (которые измеряют способность нерва посылать электрический сигнал), биопсия мышц (используется для диагностики многих нервно-мышечных расстройств) и другие анализы крови.
Скрининг новорожденных на СМА проводится во всём мире. По состоянию на январь 2021 года только в США скрининг на СМА проводится в 39 штатах, что составляет 86% всех младенцев, родившихся в США. Скрининг новорожденных способствует раннему выявлению младенцев с СМА и, следовательно, раннему назначению лечения. Младенцы, выявленные с помощью скрининга новорожденных с СМА, срочно направляются на подтверждающее тестирование, обсуждение методов лечения и ухода. Раннее лечение до появления симптомов обеспечивает наилучшие результаты. Скрининг новорожденных не выявит 3-5% младенцев с СМА из-за наличия точечной мутации в гене SMN1. У этих младенцев будут прогрессировать симптомы, и им потребуется быстрая диагностика и лечение.
Какова частота СМА?
Составляет частота СМА. СМА одинаково поражает женщин и мужчин. Скрининг новорожденных облегчает раннее выявление младенцев с СМА и, следовательно, раннее начало лечения.
Частота СМА составляет примерно 1 на 10 000 живорождений. СМА одинаково поражает женщин и мужчин. Скрининг новорожденных облегчает раннее выявление младенцев с СМА и, следовательно, раннее начало лечения. Младенцы, выявленные с помощью скрининга новорожденных с СМА, срочно направляются на подтверждающее тестирование, обсуждение методов лечения и ухода. Раннее лечение до появления симптомов обеспечивает наилучшие результаты.
Существуют ли методы лечения СМА?
Полного лечения СМА не существует. Терапия заключается в устранении симптомов и предотвращении осложнений.
В декабре 2016 года Управление по контролю за продуктами и лекарствами США одобрило нусинерсен (Spinraza™) в качестве первого препарата, одобренного для лечения детей и взрослых с СМА. Препарат вводят путем инъекции в жидкость, окружающую спинной мозг. Он предназначен для увеличения выработки полноразмерного белка SMN, который имеет решающее значение для поддержания двигательных нейронов. Польза лучше всего документирована у младенцев и детей младшего возраста, особенно при раннем начале лечения. Несколько других методов лечения находятся на поздних стадиях разработки и могут стать доступными для пострадавших людей в ближайшем будущем.
В мае 2019 года Управление по контролю за продуктами и лекарствами США одобрило генную терапию onasemnogene abeparovec-xioi (Zolgensma™) для детей младше 2 лет с СМА в младенческом возрасте. Onasemnogene abeparvovec-xioi — это генная терапия, которая доставляет полностью функциональную копию человеческого гена SMN1 в клетки двигательных нейронов-мишеней с помощью вирусного вектора AAV9. Однократное внутривенное введение препарата приводит к увеличению содержания белка SMN во всех клетках, включая двигательные нейроны.
В августе 2020 года Управление по контролю за продуктами и лекарствами США одобрило пероральный препарат рисдиплам (Эврисди) для лечения пациентов в возрасте двух месяцев и старше со СМА. Рисдиплам является первым перорально вводимым препаратом, одобренным для лечения СМА. Его механизм действия также заключается в модификации сплайсинга мРНК SMN2, что приводит к увеличению количества белка SMN.
Физиотерапия, трудотерапия и реабилитация могут помочь улучшить осанку, предотвратить неподвижность суставов и замедлить мышечную слабость и атрофию. Упражнения на растяжку и укрепление могут помочь уменьшить контрактуры, увеличить диапазон движений и улучшить кровообращение. Некоторым людям требуется дополнительная терапия при затруднениях с речью и глотанием. Вспомогательные устройства, такие как опоры или брекеты, ортопедические изделия, синтезаторы речи и инвалидные кресла, могут быть полезны для улучшения функциональной независимости.
Правильное питание и калории необходимы для поддержания веса и силы, избегая при этом длительного голодания. Людям, которые не могут жевать или глотать, может потребоваться введение питательной трубки. Неинвазивная вентиляция легких ночью может улучшить дыхание во время сна, и некоторым людям также может потребоваться вспомогательная вентиляция легких в течение дня из-за мышечной слабости в области шеи, горла и грудной клетки.
Каков прогноз при СМА?
Прогноз варьируется в зависимости от типа СМА. Некоторые формы СМА приводят к летальному исходу без лечения.
Люди с СМА могут казаться стабильными в течение длительного периода времени, но не следует ожидать улучшения без лечения.
Какие исследования СМА проводятся?
Национальный институт неврологических расстройств и инсульта (NINDS), входящий в состав Национальных институтов здравоохранения (NIH), проводит фундаментальные, трансляционные и клинические исследования SMA в лабораториях NIH, а также поддерживает исследования посредством грантов крупным медицинским учреждениям по всей стране.
Клеточные и молекулярные исследования направлены на понимание механизмов, которые вызывают дегенерацию двигательных нейронов.
Ученые проанализировали ткани человека и разработали широкий спектр модельных систем на животных и клетках для изучения процессов заболевания и ускорения тестирования потенциальных методов лечения. Среди этих усилий:
Было показано, что генная терапия и специфические препараты останавливают разрушение двигательных нейронов и замедляют прогрессирование заболевания у мышиных моделей и индивидуумов с СМА. NINDS поддержала исследования по созданию этих методов и проложила путь к клиническим испытаниям у пациентов. Клинические испытания генной терапии при СМА продолжаются.
Модели СМА на животных представляют собой важнейшие инструменты для обнаружения и разработки новых методов лечения СМА. Ученые разработали модели рыбок Данио, мышей и свиней, включая модели менее тяжелой СМА 2-го и 3-го типов, которые могут значительно помочь в определении новых терапевтических мишеней и возможных методов лечения. Ученые, поддерживаемые NIH, собрали лонгитюдные данные о предсимптомных или недавно диагностированных детях с СМА 1, 2 или 3 типов и их здоровых братьях и сестрах. Цель этого исследования - предоставить консультации и просвещение родителей о возможных возможностях клинических испытаний.