Введение
Талассемии – группа наследственных, генетических заболеваний крови, при которых нарушается синтез нормального гемоглобина (Hb). Талассемии изначально были распространены в районах неблагоприятных по заболеваемости малярией: страны Средиземноморья, Ближнего Востока, Закавказье, страны Юго-Восточной и Средней Азии. Активная миграция коренного населения данных стран привела к распространению талассемий по всему миру, в том числе России.
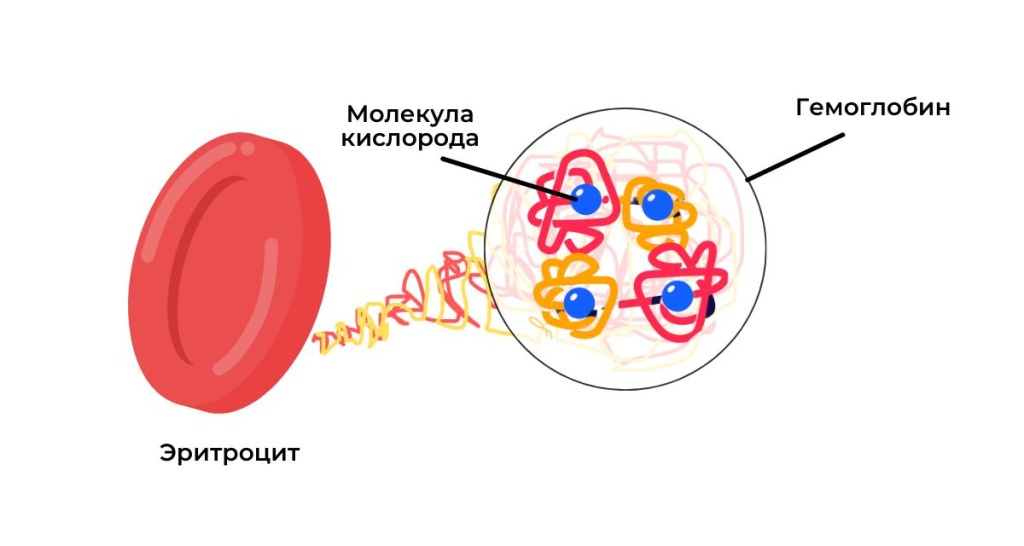
Гемоглобин сложный белок в составе эритроцитов (красных кровяных клеток), который обеспечивает транспортировку кислорода от легких к тканям и органам. Каждая молекула гемоглобина состоит из гема - железосодержащей небелкой части, и белка глобина, который представлен α, β, δ и γ-цепями. По мере взросления человека молекула гемоглобина изменяет структуру за счет смены глобиновых цепей. У взрослого здорового человека 96-98% эритроцитов содержат гемоглобин HbA. Он состоит из двух α- и двух β-цепей. Нарушение количества глобиновых цепей приводит к потере свойств гемоглобина и развитию тяжелых заболеваний.
Талассемия - описание:
β-талассемия наследственное заболевание, вызванное мутациями гена β-глобина HBB, расположенного на 11-й хромосоме. Синтеза β-глобиновых цепей кодируют два бета-глобиновых гена (β/β). Тяжесть клинических проявлений зависит от степени повреждения генов. Если у человека мутация затронула только один ген, синтез β-цепей снижается незначительно меньше нормы (β++). В этом случае заболевание протекает в легкой форме или не проявляется совсем (малая форма β-талассемии (thalassemia minor) или здоровый носитель). При возникновении мутаций в обоих генах синтез β-глобиновых цепей существенно снижается (β+) или полностью прекращается (β0). При повреждении двух генов бета-талассемия протекает в тяжелой форме– промежуточная и большая (thalassemia major, анемия Кули) формы заболевания.
Талассемия - cимптомы
Для бета-талассемии характерны следующие изменения:
Недостаток β-глобиновых цепей приводит к накоплению α- цепей. Избыток α - глобина повреждает костный мозг, нарушает кроветворение и приводит к гемолизу эритроцитов.
Неэффективный процесс образование эритроцитов стимулирует повышенное всасывание железа из пищи. Патологическое накопление железа повреждает печень, сердце, эндокринные железы. Развиваются сопутствующих заболеваний данных органов.
Что бы компенсировать нехватку эритроцитов костный мозг выходит за пределы костной ткани, образуя псевдоопухоли. Разрастание костного мозга приводит к утолщению и деформации костей, сдавлению сосудов и нервных волокон.
Увеличение селезенки происходит из-за образования дополнительных очагов кроветворения. Селезенка начинает помогать костному мозгу создавать новые эритроциты, при этом снижается количество тромбоцитов и лейкоцитов. Спленомегалия может приводить к летальному исходу из-за разрыва селезенки.
Заболевание ухудшает качество жизни и сокращает ее продолжительность за счет развития многочисленных осложнений и сопутствующих заболеваний органов (анемическое поражение сердца, тромбозы, остеопозоз, спленомегалия, трофические язвы, хроническая почечная недостаточность). Глубокая анемия приводит к отставанию в физическом и половом развитии. Нервно-психическое развитие остается в норме.
Наследование:
Бета-талассемия наследуется по аутосомно-рецессивному механизму. Ребенок будет болен, если унаследует от каждого из родителей по дефектному гену. Наследование заболевания не зависит от пола, поэтому болезнь развивается и у мальчиков, и девочек с одинаковой вероятностью. Частота встречаемости бета-талассемии по миру составляет 1 :100 000. В странах малярийного пояса этот показатель может быть выше.
Талассемия - лечение
Больные малой формой бета-талассемии и здоровые носители талассемии не требуют лечения. При промежуточной и большой формах бета-талассемии проводится симптоматическое лечение. Пожизненное переливание крови и применение железовыводящих препаратов утомительно и финансовозатратно для больных.